ChIP-seq i mapowanie sekwencji
To zadanie jest wariacją na temat zadania ChIP-seq i czynniki transkrypcyjne. Jeśli już je rozwiazałeś/aś i pamietasz, o co w nim chodziło, możesz ominąć dwa akapity wstępu.
Z genów, zapisanych na DNA powstaje mRNA, na którego bazie następnie konstruowane jest białko. Białka nie są potrzebne wszyskie naraz i w tej samej ilości - dlatego do DNA przyczepiają sie różne białka, zwane czynnikami transkrypcyjnymi, które pomagają regulować proces przepisywania DNA na mRNA. Jest to bardzo istotny proces, chociażby dlatego, ze często jego rozregulowanie jest przyczyną chorób, wiec zależy nam na tym, żeby jak najlepiej go poznać. Pomagają w tym eksperymenty zwane #ChIP-seq.
Eksperyment ChIP-seq w skrócie wykonuje sie następująco: chcemy sprawdzić, gdzie na DNA siedzi jakiś konkretny czynnik transkrypcyjny; bierzemy próbkę mnóstwa komórek; wyciagąmy z nich DNA razem z przyczepionymi czynnikami; szatkujemy DNA na kawałki; dostajemy w ten sposób mieszankę krótkich fragmentów, przy czym do niektórych wciąż przyczepione są czynniki transkrypcyjne; następnie wrzucamy przeciwciała, czyli cząsteczki specjalnie zaprojektowane tak, żeby przyczepiały sie do tego konkretnego czynnika, który chcemy zbadać; do przeciwciał były przyczepione magnesy, dzięki czemu możemy je wyciagnąc z powrotem, razem z nimi czynniki, a razem z czynnikami kawałki DNA; ostatnim krokiem jest oczyszczenie tego, co wyciągnęliśmy, z przeciwciał i czynników transkrypcyjnych i wrzucenie pozostałych fragmentów DNA do sekwenatora, dzięki czemu poznamy ich sekwencje. Właściwie nie jest to ostatni krok, bo z samych krótkich fragmentów sekwencji nic nie wynika. Ostatnim jest analiza bioinformatyczna wyniku.
W pliku odczyty.fasta znajduje sie wynik działania sekwenatora, czyli sekwencje, które udało nam sie wyciągnąć z próby. Spodziewamy
się, że są to sekwencje z miejsc, do których przyczepiony był czynnik transkrypcyjny.
W pliku genom.fasta jest cała sekwencja genomu, z którego te odczyty pochodzą. Twoim zadaniem będzie sprawdzenie na podstawie tych danych, ile czynników transkrypcyjnych siedziało na badanym DNA.
Uwaga nr 1. Ponieważ eksperymenty ChIP-seq (jak większość eksperymentów) nie jest idealna, niestety zawsze z próby wyciągnie się też trochę śmieci. Dlatego sam fakt, że znaleźliśmy fragment z jakiegoś miejsca jeszcze nie świadczy, że w tym miejscu rzecywiście był czynnik transkrypcyjny. Przyjmijmy, ze jeżeli z jakiegoś miejsca znalazło się 5 odczytów to będziemy uważa
ć
, że to mało prawdopodobne że znalazły się tam przez pomyłkę i uznamy, że rzeczywiście czynnik transkrypcyjny w tym miejscu był.
Uwaga nr 2. Jeśli rozwiazywałaś/eś zadanie ChIP-seq i czynniki transkrypcyjne, pamiętasz, że polecenie było podobne, ale jako dane wejściowe podany był plik, w którym policzone już było, ile fragmentów z sekwenatora znalazło się w każdym z fragmentów genomu. Tym razem dostajemy same odczyty i sami musimy sobie poradzić ze znalezieniem, gdzie są w genomie.
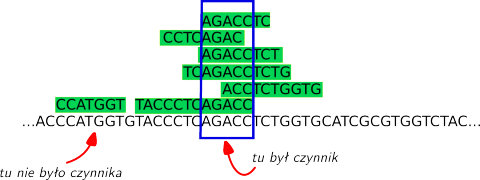
Przykład: w pliku odczyty.fasta znajdowały się sekwencje, zaznaczone poniżej na zielono, zaś w pliku genom.fasta sekwencja zapisana pod nimi. Znaleźliśmy, z jakich miejsc w genomie pochodziły zsekwencjonowane fragmenty. Fragment CGATGGT jest zupełnie sam w tym miejscu, przyjmujemy więc, że znalazł się w próbce przez przypadek. Z kolei na fragmencie AGACC znajduje się aż 5 lub więcej fragmentów - uznajemy więc to za istotne statystycznie i przyjmujemy, że w tym miejscu rzeczywiście był czynnik transkrypcyjny.
| Załącznik | Wielkość |
|---|---|
| 100.12 KB | |
| 17.83 KB |
- Zaloguj się albo zarejestruj aby dodać rozwiązanie
-

